

Tra le malattie che possono colpire il cervello vi è la Corea di Huntington. Questa è una malattia neurodegenerativa di importante significato clinico. Qual è la sua genetica, come si eredita e quali sono i sintomi?
Indice dell'articolo
Cause della Corea di Huntington
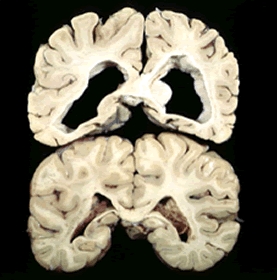
La Corea di Huntington è purtroppo genetica. È causata da una mutazione ereditaria che porta ad un anomalo “allungamento” di un gene, conosciuto come gene HD. Nella maggior parte dei casi quindi un paziente affetto da Huntington trasmette la malattia ai suoi figli, e che l’insorgenza della malattia può variare drasticamente.
Circa il 3% dei soggetti affetti invece non ha ereditato la malattia dai genitori; comunque anche in questi casi la mutazione del gene è avvenuta durante lo sviluppo fetale.
Il normale gene HD prevede la produzione della proteina huntingtina di cui però ancora non si conosce bene la funzione; tutto quello che si sa di questa proteina è che è maggiormente diffusa nel cervello e che interagisce con importanti attività cellulari. Nei casi di gene HD mutato, la proteina huntingtina anomala risultate causa disfunzioni neuronali, atrofia generalizzata all’interno del cervello e morte dei neuroni, che sono le cellule cerebrali.
Perché si chiama Corea di Huntington?
Il significato del nome Corea di Huntigton deriva dal greco χορεία (chorèia) e dal latino “chorea”. Con questa parola si intende un quadro clinico caratterizzato da movimenti rapidi ed incontrollati che hanno un’origine neuropatologica.
Sintomi della Corea di Huntington
Questo ci fa intendere chiaramente che tra i sintomi della malattia di Huntington vi sono, appunto, degli spasmi non ripetitivi e non periodici dei muscoli volontari che possono anche interferire con le attività quotidiane e che col progredire dalla malattia possono poi anche colpire i muscoli involontari e rendere il paziente non autosufficiente.
Ai problemi muscolari si accompagnano i disturbi cognitivi che possono insorgere molto presto, anche se la perdita del linguaggio arriva sono verso la fine del decorso della malattia. Al contrario, i disturbi comportamentali scompaiono con l’avanzare della Corea di Huntington e possono includere la tendenza all’isolamento, l’aggressività, le deviazioni sessuali, l’apatia e l’aumento dell’appetito.
Quando questi sintomi scompaiono il soggetto inizia a perdere peso repentinamente, diminuisce il sonno e aumenta il mutismo, fino ad arrivare a casi di schizofrenia, cambi della personalità e psicosi.
Quando insorge?
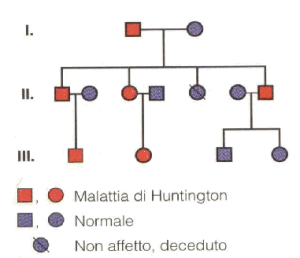
La Corea di Huntington ha vari livelli di espressività. Come già anticipato, l’esordio e lo sviluppo della malattia variano in base all’entità dell’allungamento del gene HD; questo aumenta passando di generazione in generazione, ed è per questo che purtroppo i figli malati tendono ad avere i primi sintomi più presto rispetto al loro genitore.
Nonostante vi siano stati rari casi di pazienti che hanno cominciato a sviluppare i primi sintomi a 50 anni, l’età media di insorgenza è compresa tra i 35 e i 44 anni. Il decorso della malattia lungo circa 15-18 anni, e questo porta ad una età media di decesso intorno ai 55 anni. Nei casi di età d’insorgenza precoce per un genitore, un figlio malato potrebbe avere i primi disturbi comportamentali già nella tarda infanzia.
Cura per la malattia di Huntigton
Purtroppo non esiste alcun tipo di cura per i malati di Corea di Huntington. L’unico trattamento attualmente in uso è quello che prevede la somministrazione di farmaci in grado di controllare e ridurre i sintomi comportamentali e neurologici, come la tetrabenazina.
È possibile però predire la malattia tramite test genetici prenatali che però necessitano di una conferma grazie all’analisi di un’altra persona affetta della famiglia.
Alessandra Spaziano
Fonti:
- Thompson & Thompson – Genetica in Medicina
- http://www.lirh.it/it