

Indice dell'articolo
Kuru: un’epidemia rituale

Il Kuru è una malattia neurodegenerativa infettiva, scoperta in seguito ad un picco epidemico negli anni ’50 e ’60 del secolo scorso, che portò al decesso di circa un migliaio di indigeni Fore, soprattutto tra donne e giovani.
I Fore sono una popolazione indigena del territorio della Papua Nuova Guinea dedita all’agricoltura e all’allevamento che oggigiorno conta circa 20,000 individui. Sebbene la loro identità culturale sia sempre più contaminata dalle popolazioni vicine e dai missionari Europei, la religione dei Fore era fino al secolo scorso prevalentemente politeistica, con un’importante componente di spiritismo e sciamanesimo.
Secondo le loro passate credenze ogni individuo possiede 5 anime e una di queste, l’auma, può essere trasmessa ai propri familiari dopo la morte se questi si fossero cibati delle carni del defunto: in particolare donne e bambini avrebbero dovuto mangiare cervello e visceri, mentre ai maschi adulti si riservavano i muscoli.
Questa pratica, ormai desueta nella popolazione Fore, fu la principale causa della gravissima epidemia degli anni ’50-’60. Sebbene con trend decrescente, i casi di kuru si protrassero fino agli anni ’70 anche dopo l’abbandono del cannibalismo rituale, e ciò è stato attribuito al lungo periodo di incubazione della malattia, uguale a circa 10-50 anni.
Una malattia misteriosa
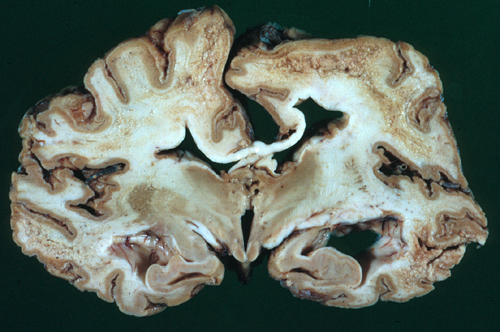
Gli scienziati europei che studiarono i cadaveri di coloro che si erano ammalati del terribile morbo rilevarono un’interessante anomalia cerebrale: in particolare il tessuto nervoso appariva rammollito e spugnoso.
L’aspetto macroscopico suggerì che la misteriosa malattia potesse appartenere al gruppo delle encefalopatie spongiformi, di cui faceva parte la già nota malattia di Creutzfeldt-Jakob. Successivamente si riuscì a isolare il probabile agente infettivo responsabile, che tuttavia sembrava essere composto solo da una singola proteina, il che appariva assurdo agli studiosi del tempo. A questa nuova entità venne dato il nome di Prione, acronimo di “PRoteinaceus Infective ONly Particle”, ovvero “particella infettiva esclusivamente proteica”.
Prioni: più elementari dei virus
Secondo le convinzioni degli scienziati dei primi anni del ‘900, un qualsiasi organismo capace di riprodursi avrebbe dovuto avere necessariamente un genoma composto da DNA o RNA, cioè le molecole fondamentali della trasmissione genetica.

Anche i virus, i microrganismi più semplici conosciuti, non possono prescindere dagli acidi nucleici per la propria diffusione: sappiamo infatti che somministrare solo la componente proteica del virus, privandolo del proprio genoma, non è sufficiente a causare e protrarre l’infezione (su questo principio infatti si basano alcuni vaccini).
Questo postulato venne messo in discussione quando si scoprirono i Prioni, semplici proteine che però si comportavano come veri e propri organismi infettivi e quindi una volta inoculati nell’ospite si riproducevano e potevano produrre contagio.
La proteina prionica (PrP-c) è un normale prodotto del nostro genoma, in particolare del gene PrP presente sul cromosoma 20. Tale proteina può subire però delle modificazioni che la rendono responsabile delle lesioni cerebrali.
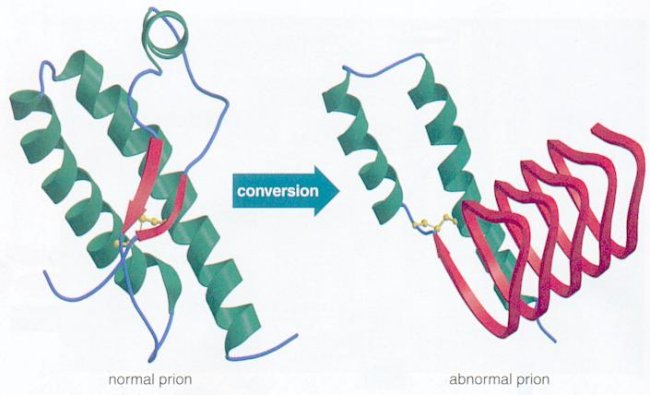
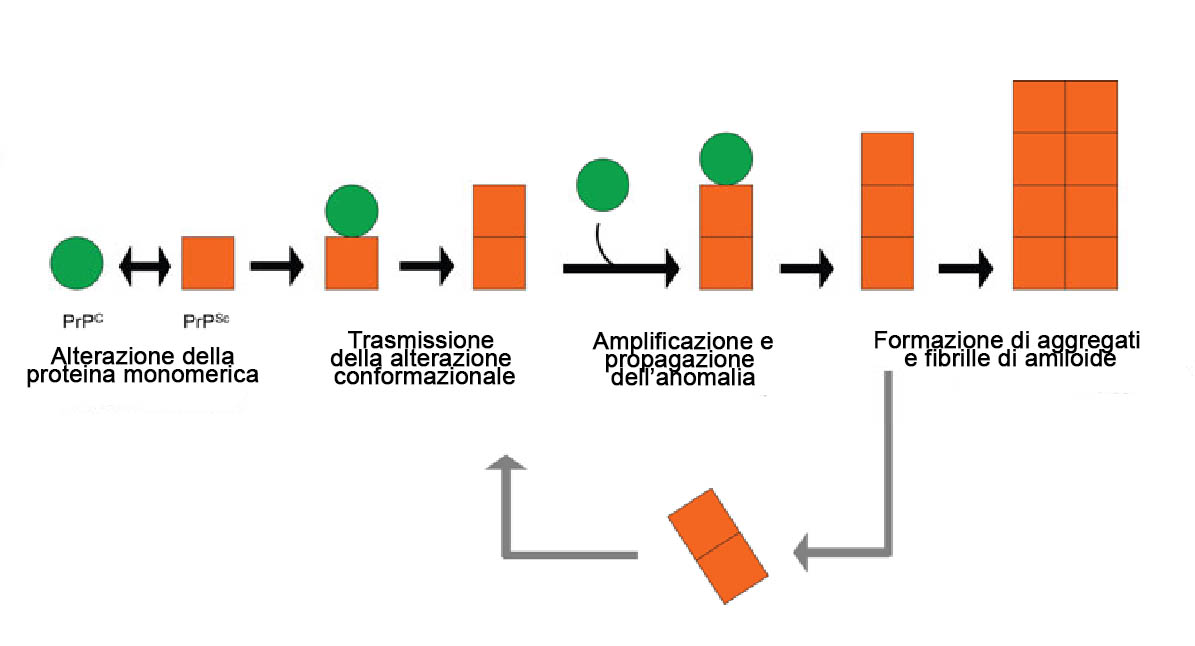
Queste modificazioni possono essere causate o da mutazioni genetiche o da interazione con la PrP-Sc, cioè la versione anomala della proteina PrPc. La PrP-Sc infatti ha la peculiare caratteristica di poter indurre modifiche conformazionali alle proteine normali, alterandone la struttura. In altre parole è come se la proteina malata “infettasse” le proteine sane rendendole a loro volta “contagiose”.
Anatomia patologica
Questa reazione a catena porta alla formazione di aggregati proteici chiamati placche amiloidi (presenti in molte altre malattie neurodegenerative come l’Alzheimer) che si accumulano nel tessuto cerebrale e provocano morte dei neuroni. Nel caso del kuru il prione si trasmetteva da individuo ad individuo tramite l’atto di mangiare il cervello dei malati, essendo tale organo il più ricco di agente infettivo.
Le malattie causate da prioni prendono complessivamente il nome di encefalopatie spongiformi e se ne riconoscono alcuni tipi oltre al Kuru, tra cui le più importanti sono la malattia di Creutzfeldt-Jakob e l’insonnia familiare fatale. Non tutte le malattie da prioni però sono infettive come la kuru, essendo in molti casi ereditarie e genetiche.
Clinica del Kuru e delle altre malattie da prioni
Il danno causato dall’accumulo di proteina prionica è diversamente localizzato nel cervello a seconda del tipo di malattia prionica in esame. In base della sede cerebrale colpita si avrà la predominanza di un certo tipo di sintomi e segni clinici.
Nel caso del Kuru le lesioni sono principalmente cerebellari (cioè del cervelletto) e più precisamente del paleocerebellum, l’area del cervelletto specializzata nella coordinazione dei movimenti. Il danno in questa sede comporta infatti atassia, disturbo neurologico caratterizzato da alterazione dell’equilibrio e incoordinazione motoria.
A questo si aggiunge un vistoso tremore (“Kuru” in lingua fore significa appunto “tremore”), decadimento della coscienza e coma fino alla morte, che insorge in media 12 mesi dopo l’esordio dei sintomi.
Questo rappresenta un decorso paradigmatico di quasi tutte le encefalopatie spongiformi, sebbene ogni sottotipo abbia le proprie peculiarità. Purtroppo ad oggi non è presente nessuna terapia accertata che permetta di invertire o arrestare l’inesorabile e funesta progressione della degenerazione cerebrale.
La malattia di Creutzfeldt-Jakob (CJD), malattia da prioni più celebre e diffusa rispetto al kuru, comprende diversi forme:
- familiare, ereditaria, causata da una mutazione del gene PrP che predispone al precoce sviluppo della malattia.
- sporadica, senza associazione familiare, senza causa nota e con esordio senile.
- iatrogena, ormai scomparsa, dovuta al fatto che in passato alcuni farmaci per malattie ormonali venivano purificati da estratti di cervello animale.
- variante (vCJD), comunemente detta “malattia della mucca pazza“, si diffuse soprattutto negli anni ’90 tramite il consumo di carne bovina infettata dal prione; dimostra un’insorgenza giovanile e una prevalenza dei disturbi comportamentali su quelli propriamente neurologici.
L’insonnia familiare fatale è una rara malattia da prioni ereditaria che colpisce primariamente il talamo provocando insonnia totale e deficit della memoria; l’insorgenza è giovanile e il decorso è infausto analogamente alle altre forme di encefalopatie spongiformi.
Prospettive di ricerca e conclusioni
In uno studio del 2017 pubblicato sul Journal of Neurology si sostiene che sia possibile diagnosticare precocemente la malattia di Creutzfeldt-Jakob attraverso analisi del liquor cerebrospinale e delle urine, e di conseguenza instaurare una terapia con doxiciclina, comune antibiotico che pare avere effetti benefici sulla patologia; tuttavia i risultati in quanto a miglioramento della sopravvivenza e della qualità della vita non sembrano particolarmente promettenti.
Nelle malattie prioniche infettive è stato fodamentale il ruolo della prevenzione, attraverso cui è stato possibile ridurre la diffusione dei prioni mediante il controllo delle possibili fonti di contagio come alimenti, trapianti e materiali sanitari infetti.
Le malattie neurodegenerative rappresentano forse la sfida più ardua per i ricercatori, ma è necessario non perdere la speranza poiché anno dopo anno il progresso scientifico dimostra che per superare certi limiti apparentemente invalicabili serve solo tempo, dedizione e fiducia nella ricerca.
Antonio Spiezia
Bibliografia
Abbas A.K., Aster J.C., Kumar V., Robbins e Cotran, Le basi patologiche delle malattie (IX edizione), Seregno, Edra Masson, 2015
Alberts B., et al., Biologia molecolare della cellula (V edizione), Bologna, Zanichelli, 2009
Anastasi G., et al., Trattato di Anatomia umana, Milano, edi-ermes, 2010
Barone P., Brunetti A., Cappabianca P., Filla A., Gangemi M., Maiuri F., Santoro L., Spaziante R., Sistema nervoso – Neurologia – Neurochirurgia – Neuroradiologia, Napoli, Idelson – Gnocchi , 2012
Sitografia
https://en.wikipedia.org/wiki/Fore_people
https://en.wikipedia.org/wiki/Kuru_(disease)
https://en.wikipedia.org/wiki/Prion
https://en.wikipedia.org/wiki/Creutzfeldt%E2%80%93Jakob_disease
http://n.neurology.org/content/70/15/1272
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5413516/
ATTENZIONE: Le informazioni contenute in questo sito hanno puramente scopo informativo e divulgativo. Questi articoli non sono sufficienti a porre diagnosi e decisioni di trattamento e non sostituiscono mai il parere del medico. Per ulteriori informazioni contattare il proprio medico generico o specialista.